. Infections and autoimmune diseases. J Autoimmun. 2005;25 Suppl:74-80. doi: 10.1016/j.jaut.2005.09.024. Epub 2005 Nov 8.
[PMID: 16278064] [DOI: 10.1016/j.jaut.2005.09.024]
- For Patients
- Introduction to the Marshall Protocol
- Length of the MP
- Immunopathology
- Managing immunopathology
- Food and drink
- Light restriction
- Vitamin D metabolite calculator - get feedback on vitamin D results
- Therapeutic probe - best method for determining MP suitability
- Starting the Marshall Protocol
- Resources for patients
- Marshall Protocol summary
- Palliative vs. curative treatments
- Patient interviews on Bacteriality
- Non-MP treatments
- Glossary
- The Protocol
- Marshall Protocol summary
- Publications and presentations
- Science behind immunopathology
- Science behind olmesartan (Benicar)
- Safety of olmesartan
- Resources for physicians
- Vitamin D metabolite calculator - get feedback on vitamin D results
- Therapeutic probe - best method for determining MP suitability
- Physicians' concerns about the MP
- Notice for emergency medical personnel

- The Pathogenesis
- Publications and presentations
- Science behind Marshall Pathogenesis
- Microbes in the human body
- Successive infection and variability in disease
- Autoimmune theory of disease
- Science behind vitamin D
- Metabolism of vitamin D and the VDR
- Th1 Spectrum Disorder - how chronic inflammatory diseases are related
- Transmission of bacteria and onset of chronic disease
- Evolutionary perspective on disease
- Incidence and prevalence of disease
- Evidence that chronic disease is caused by pathogens
- Diseases
- Foundation
- Related Sites


Table of Contents
Genetic predisposition to disease
Many researchers have long argued that most chronic diseases are caused by humans' genetic predisposition for a condition. However, despite ambitious efforts, there is substantial evidence that chronic diseases are not caused by human genes. Studies of monozygotic (fraternal) twins are particularly damning. The high percentage of disease-discordant pairs of monozygotic twins demonstrates the central role of environmental factors (like microbes) in the cause of autoimmune diseases, to say nothing of similar data about other inflammatory diseases such as cancer.1)
According to the Marshall Pathogenesis, humans accumulate a plethora of pathogenic bacteria during their lifetimes, and it is the genetic mutations and disruption of key transcriptional pathways which result from active infection that play a major role in what is commonly thought of as “genetic susceptibility.”
If anything, we don't really know how to read the genome and it can't tell us very much right now. So what's the ethical debate about?… We couldn't even be certain from my genome what my eye color was. Isn't that sad? Everyone was looking for miracle 'yes/no' answers in the genome. “Yes, you'll have cancer.” Or “No, you won't have cancer.” But that's just not the way it is…. [The Human Genome Project has had] close to zero [medical benefit] to put it precisely…. We have, in truth, learned nothing from the genome other than probabilities. How does a 1 or 3 percent increased risk for something translate into the clinic? It is useless information.
J. Craig Venter, Der Spiegel interview
Human genetics as a cause for chronic disease
The bulk of the human genome was first fully sequenced in the year 2003. For some, the “Genomic Era” - as it has been called 2) - represents the future of medical therapies for chronic disease. Using rapid throughput genomic sequencing technology, researchers have conducted studies comparing the genetic makeup of people suffering from a given chronic disease, sometimes as many as 1,000 or more, to healthy controls. Researchers have long hoped that these studies will produce a list of which human genes cause which chronic diseases and allow for true genomically-based “personalized medicine” including the administration of genomic-based designer drugs as well as gene therapies.3)
Optimism that this avenue of research will improve clinical outcomes has always been high and is epitomized by this 2001 paper in JAMA:
It should be possible to identify disease gene associations for many common illnesses in the next 5 to 7 years.
Francis Collins, et al. 4)
Failed promise of human genetic research
It may yet be too early to call human genomic research an unqualified failure, but certain researchers have noted a distinct lack of results, and limited progress in the genetic analysis of common diseases has been widely acknowledged:5) 6) 7)
- “Expectations regarding the benefits of genetic research” have been “overly enthusiastic.”8)
- “Much effort has been expended on research into their causes, with the aim of predicting who will be affected or preventing effects before they arise, but progress has been halting at best.”9)
- “Given the continuing difficulty of identifying genes for complex disorders in a robust, replicable manner, and the extensive resources devoted to this effort, it is becoming increasingly important to analyze the relative benefits of genomics research for public health applications and for the understanding of disease pathogenesis.”10)
- “Heritable causes of complex diseases remain largely elusive, despite tremendous efforts to understand them…. Technological excellence in genomics does not automatically lead to benefits in human health.”11)
In a striking image from a 1999 lecture, Francis Collins of the US National Human Genome Research Institute described a hypothetical consultation in 2010, in which a 23-year old man has a high concentration of cholesterol identified during screening and undergoes extensive genetic testing.12) The scenario listed eight genetic variants which at the time were thought to contribute to disease. What has been the fate of these genetic variants? According to Smith: “With few exceptions, later [more conclusive] evidence suggests that these variants are related to much smaller increased risks of disease, if any,” and would not be of value during a medical consultation.13)
We know that diseases cluster in families. In some diseases, the risk might be two or three times higher than normal, or 30 times higher, for a relative of someone with a disease. But when we do these genomic studies, we find maybe a 50 percent increase in risk. That gap is what’s missing.
Teri Manolio, Director of the National Human Genome Research Institute’s Office of Population Genomics, discussing missing heritability in a 2009 Wired interview
Conventional explanations for why genetic studies have failed
In accounting for the failure to identify the genes which supposedly cause chronic disease, there seem to be two popular explanations:
- The sheer complexity of the human genome does not lend itself to ready understanding of which genes cause which diseases.
- Disease is caused by the interaction between genes and the environment.
However, these reasons do not account for the fact that few, if any, “high penetrance” genes have been identified. In the words of Hammiki et al., “The present flood of genetic and genomic data and references to genomic medicine might give the impression that” these are solid and likely explanations, but “many studies point to a predominantly environmental causation of disease.”14)
Diseases
Most common gene variants that are implicated by such studies are responsible for only a small fraction of the genetic variation that we know exists. This observation is particularly troubling because the studies are largely comprehensive in terms of common single-nucleotide polymorphisms (SNPs), the genomic markers that are genotyped and with which disease associations are tested. Researchers have finding the biggest effects that exist for this class of genetic variant, and common variation is packing much less of a phenotypic punch than expected. Indeed, the reality of genome-wide association studies has been disappointing in the case of at least several prominent diseases.
Cancer
Even researchers studying a high-priority, frequently studied disease such as cancer have failed to produce a telltale genetic association.
Genetic mutations are rampant in cancerous tissue: compared to tissue from a healthy age-matched control, a lung cancer patient may have as many as 50,000 genetic mutations.15) However, among the inherited cancer syndromes with no readily discernible environmental influence, it is thought that only 1-2% are caused by high penetrance (high-risk) human genes.16)
When diseases are not inherited in Mendelian ratios - somewhere between 0.01% and 0.001% of live births suffer from Mendelian diseases - geneticists have argued that those diseases are driven by effects reflecting a complicated interplay of human genes. But if a disease is a “genetic disease,” it should show very distinct patterns among twins, especially monozygotic (identical) twins. Because monozygotic twins are virtually identical in genetic makeup, the monozygotic twin concordance for such genetic diseases should be virtually 100 percent. For most diseases, this is not the case.17)
Monozygotic twins share the same genotype because they are derived from the same zygote. However, monozygotic twin siblings frequently present many phenotypic differences, such as their susceptibility to disease [italics added] … Recent studies suggest that phenotypic discordance between monozygotic twins is at least to some extent due to epigenetic [changes in gene expression caused by mechanisms other than changes in the underlying DNA sequence] factors that change over the lifetime of a multicellular organism.
P. Poulsen, et al. 18)
One study found that genetic concordance rates for identical twin pairs by age 75 years were only 11% for colorectal cancer, 13% for breast cancer and 18% for prostate cancer, and these were lower in dizygotic twins (5%, 9% and 3%, respectively).19) These low concordance rates agree with other data on cancer incidence and provide little support for strong heritable effects in cancer.20)
Infection with a bacteria called Helicobacter pylori has been widely recognized as the cause of stomach ulcers, but it is now thought to also cause stomach cancer.21) It's worth noting that the causative role H. pylori in stomach ulcers was not identified even though researchers were able to cultivate this bacterium in the laboratory and to see it under the microscope in gastric biopsies.
Persistent human papillomavirus infections have been recognized as the major cause of cervical cancer and may play a role in some cancers of the anus, vulva, vagina, and penis.22) Evolutionary biologist Paul Ewald has argued that infectious causation has often been accepted belatedly throughout the history of medicine and, if decades long trends are anything to go by, mainstream medicine will ultimately agree that the majority of cancers are caused by pathogens:
Back in 1975, mainstream medicine agreed that about 0.1% of human cancer cases were caused by pathogens. When it came to the rest of cases, their view was that they were probably caused by a combination of inherited predispositions and mutagens. Then in 1985, the percentage of cancer cases they tied to pathogens was 3%, and they continued to make the same argument about the remaining cases. In 1995 the percent of pathogen-induced cancer cases was accepted to be around 10%. Now, we’re at 20%. Still, mainstream medicine contends that the other 80% of cases do not have an infectious cause, but the question is – do you believe them anymore? In this sense, the clarity of hindsight can help a lot. Between evolutionary instinct and plain common sense we can view the issues of pathogens and cancer much more effectively.
Paul Ewald, PhD, Bacteriality.com
In a 2000 paper co-written with Ewald, Cochran and Cochran trace the long and seemingly inevitable acceptance of chronic diseases as being caused by infectious agents rather than genetic predisposition.23)
Schizophrenia
In 2009, a press conference at the World Conference of Science Journalists, triumphantly unveiled three large studies of the genetics of schizophrenia.24) Press releases from five American and European institutions celebrated the findings, one using epithets like “landmark,” “major step forward,” and “real scientific breakthrough.” The media coverage for these papers was likewise triumphal, talking about breakthroughs and a discovery that could lead to new treatments for millions with the illnesses. One exception was a Reuters article that said, “as many as 30,000 different gene variations may underlie schizophrenia and bipolar disease, meaning any kind of quick test to predict either disease is a long way off.”
It was the kind of hoopla you’d expect for an actual scientific advance. It seems to me the reports represent more of a historic defeat, a Pearl Harbor of schizophrenia research….
In the last few years gene hunters in one common disease after another have turned up a few causative variant genes, after vast effort, but the variants generally account for a small percentage of the overall burden of illness. With most common diseases, it turns out, the disease is caused not by ten very common variant genes but by 10,000 relatively rare ones.
Today it’s the turn of schizophrenia researchers to make the same discovery, though one perhaps more to be expected since schizophrenia is not good for reproduction.
Schizophrenia too seems to be not a single disease, but the end point of 10,000 different disruptions to the delicate architecture of the human brain.
Nicholas Wade, New York Times July 2009
Genes of tissue are different than blood
One of the basic assumptions underlying many genome-wide association studies has been that the genetic makeup of all an individuals’ cells is essentially the same.25) In the vast majority of genetic studies to date, researchers have assumed that in sequencing DNA isolated from blood would reveal the genetic makeup of diseased tissues as well. This supposition was convenient: except for cancer, samples of diseased tissue are difficult or even impossible to take from living patients.26)
However, recent evidence has emerged that the genes of at least some cells from the blood and tissue do not match genetically,27) meaning that ambitious and expensive genome-wide association studies may prove to have been essentially flawed from the outset.
Systematic errors in the interpretation of the genome
In a 2010 book chapter, members of the Autoimmunity Research FoundationNon-profit foundation dedicated to exploring a pathogenesis and therapy for chronic disease.'s Research Team predicted that records of the human genome almost necessarily contained microbial DNA in them.
Primers selected for most epidemiological studies are chosen without consideration for whether they might amplify DNA from the genomes of any intracellular microbes. As artist Pablo Picasso once remarked, “Computers are useless. They can only give you answers.” If a software program fails to make provision for the possibility that a metagenome might also be present, the chances of a false positive increase significantly during the process of genomic analysis. Similarities between bacterial and human genes will likely cause the analysis software to not assemble the genomic data properly. The likelihood of error is not minuscule as there is growing evidence of molecular mimicry, homology between bacterial and human proteins….
Before we can be certain that all measured SNPs and HLA haplotypes are a product of only the human genome and not the metagenome, researchers must begin to actively choose PCR primer pairs that are unlikely to amplify microbial DNA. Primers need to be certified not only to amplify a unique sequence in the human genome, but also as not likely to amplify genes from any of the thousands of bacterial and viral genomes in the metagenomic databases. Although PCR amplification usually involves more than one stage of genomic selectivity, the increasing use of arrays of RNA probes increases the likelihood that a fragment of metagenomic RNA will unexpectedly match a probe, and increases the possibility of a false-positive being signaled for the particular SNP being sought.
Amy Proal, Paul Albert, and Trevor Marshall, PhD 28)
In their 2011 publication, More Mouldy Data: Virtual Infection of the Human Genome, Langdon and Arno show that records of the human genome have become contaminated with microbial DNA. The London-based researchers found sequences from mycoplasma bacteria, an intracellular microbe, in GenBank. At least one sequence for mycoplasma was incorporated into commercial tools such as microarrays. The authors argue that there is a great need to clean up genomic databases but fear that current tools will be inadequate to catch genes which have jumped the silicon barrier.
The level of contamination and the way in which it is spreading suggests that researchers are losing the battle to eliminate it…. Most frightening of all is the possibility that Langdon and Arno may have only scratched the surface. “Having found two suspect DNA sequences, it seems likely the published “human genome” sequence contains more,” they say…. Clearly, this is a nettle that needs to be grasped quickly. That's if it's not too late already.
Bacteria are "inherited" between generations
There’s a 0.1% difference in our genomes, but there may be a 50% difference in our metagenome. If the variability is higher, you’re more likely to find a relevant signal…. It’s hard to be a prophet, but we see so much more potential in the human-other-genome than in our own genome.
Dusko Ehrlich, Project Coordinator for MetaHIT
Strong evidence indicates that pathogens play the primary role in chronic disease. Scientists have confirmed that bacteria are transmitted through a series of vectors including:
- from mother to fetus during pregnancy29)
- from father to child via sperm30)
- through social contact31)
While there is no evidence that human genes contain instructions for chronic disease, there is reason to believe that a body's unique pathogenic load, its pea soup, causes mutations in human DNA. Pea soupThe unique combination of bacterial pathogens (and co-mingling of bacterial genes) which accounts for each individual’s disease presentation. refers to the diverse and ever-changing human microbiota, a broad array of bacteria comprised of various forms and at least hundreds of species. Many of these bacteria were thought only recently never to exist in man. A 2007 study, for example, found hydrothermal vent eubacteria on a prosthetic hip joint.33)
Pathogens may cause genetic mutations
Because the bacteria which drive chronic disease are highly diverse, it makes sense that the human genetic differences observed by scientists between one person and the next are inconsistent. But, it is possible to measure the general damage pathogens do to human DNA – or, perhaps more importantly, the damage they do to the body's ability to repair DNA. These analyses show that human DNA changes in accordance with disease progression and that pathogens are plausible, if not likely, culprits.
What the researchers are seeing as changes on genes are indeed changes, but they only correlate at low levels of significance because they are due to pathogens. They are due to mutations from chronic infection. Consequently there is no causal effect - only an associative observation.
Trevor Marshall, PhD

Few seem to appreciate that one's genome can change over a lifetime of accumulating pathogens. According to Marshall Pathogenesis, many of the genetic mutations identified by Human Genome Project researchers are largely induced by bacteria and other pathogens. For example:
- A 2011 Nature Genetics study found that as many as 20 percent of sporadic autism cases can be explained by de novo mutations, also known as spontaneous gene mutations.34) A 2012 Nature study (right) found in a cohort of 78 Icelandic parent-offspring trios that genetic mutations in children become more numerous with advancing paternal age. Kong et al. concluded that these mutations may explain as many as 30 percent of autism cases.35) The effect is an increase of about two mutations per year. An exponential model estimates paternal mutations doubling every 16.5 years.
- The HLA axis genes are often cited as predisposing a person to certain kinds of diseases. Yet, there is no determinate diagnostic value to be obtained by measuring genes in the HLA axis, which include HLA-DRA, HLA-DRB and HLA-DQ. None of the HLA haplotypes causes disease 100% of the time and none cause any one disease consistently.
Only the variable influence pathogens have on DNA can explain this phenomenon.
Rather than serving as markers of particular diseases, such mutations generally mark the presence of those pathogens capable of affecting DNA transcription and translation in the nucleus.
This disease process leaves geneticists with the task of examining a perplexing number of different mutations, most of which differ so greatly between individuals that no correlations can be made between their presence and any particular illness. The mutations are nothing but genetic “noise,” induced either by random chance or by the pathogens that such researchers fail to factor into the picture.
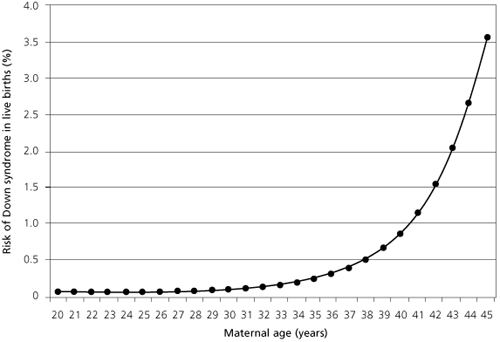
Risk of Down syndrome increases as a woman ages. The increased risk suggests involvement of Th1 pathogensThe community of bacterial pathogens which cause chronic inflammatory disease - one which almost certainly includes multiple species and bacterial forms..
Fetal development defects
Down syndrome is a disease in which one of three types of abnormal cell division involving the 21st chromosome occur. Except for a certain rare case, Translocation Down syndrome, Down syndrome is not inherited.
The risk for Down syndrome increases as a woman ages (right).36)
Descriptions of the etiology of Down syndrome usually offer that the disease occurs, because of a mistake in cell division during the development of the egg, sperm or embryo. But, these types of generalized explanations do not explain why the mistakes occur.
According to the basic principles of evolutionary theory, a disease with the prevalence of Down syndrome could not be caused by human genes.
Pathogens are one plausible explanation for the causation of fetal development defects like Down syndrome. Researchers have documented that pathogens are present in the human sperm37) as well as in the human female reproductive organs.38) The risk for producing a child with Down syndrome increase exponentially as a woman ages, something which could be attributed to the accumulation of pathogens.
Also, this:
One of the genes known to be transcribed by the Vitamin D Receptor [which controls innate immunity] is located in the region known to be responsible for Down syndrome. The function of this gene is currently unknown, but it certainly seems as though Th1 pathogens might well be involved in this most classic of “birth defects.”
Trevor Marshall, PhD
Epigenetics
Even relatively straightforward traits such as height have resisted attempts to reduce it to a particular combination of genes. In light of this shortcoming, some investigators see room for an increased focus on an alternative explanation for heritable traits: epigenetics, the molecular processes that control a gene’s potential to act. Evidence now suggests that epigenetics can lead to inherited forms of obesity and cancer. The best-studied form of epigenetic regulation is methylation, the addition of clusters of atoms made of carbon and hydrogen (methyl groups) to DNA.
Bacteria make widespread use of postreplicative DNA methylation for the epigenetic control of DNA-protein interactions. Bacteria make use of DNA adenine methylation (rather than DNA cytosine methylation) as an epigenetic signal. DNA adenine methylation is important in bacteria virulence in organisms such as Escherichia coli, Salmonella, Vibrio, Yersinia, Haemophilus, and Brucella. In Alphaproteobacteria, methylation of adenine regulates the cell cycle and couples gene transcription to DNA replication. In Gammaproteobacteria, adenine methylation provides signals for DNA replication, chromosome segregation, mismatch repair, packaging of bacteriophage, transposase activity and regulation of gene expression.39) 40)
Read more
- We Have Learned Nothing from the Genome – 2010 SPIEGEL Interview with Craig Venter
- Beyond the Genome – 2009 Wired article that describes how when scientists finished sequencing the human genome, the answers to diseases were supposed to follow. Six years later, that promise has gone unfulfilled.
- Hoopla, and Disappointment, in Schizophrenia Research – 2009 New York Times article on the failure of genomic association studies in schizophrenia research
2)
. Welcome to the genomic era. N Engl J Med. 2003 Sep 4;349(10):996-8. doi: 10.1056/NEJMe038132.
[PMID: 12954750] [DOI: 10.1056/NEJMe038132]
[PMID: 12954750] [DOI: 10.1056/NEJMe038132]
3)
, 4)
. Implications of the Human Genome Project for medical science. JAMA. 2001 Feb 7;285(5):540-4. doi: 10.1001/jama.285.5.540.
[PMID: 11176855] [DOI: 10.1001/jama.285.5.540]
[PMID: 11176855] [DOI: 10.1001/jama.285.5.540]
5)
, 13)
. Genetic epidemiology and public health: hope, hype, and future prospects. Lancet. 2005 Oct 22-28;366(9495):1484-98. doi: 10.1016/S0140-6736(05)67601-5.
[PMID: 16243094] [DOI: 10.1016/S0140-6736(05)67601-5]
[PMID: 16243094] [DOI: 10.1016/S0140-6736(05)67601-5]
6)
. A haplotype map of the human genome. Nature. 2005 Oct 27;437(7063):1299-320. doi: 10.1038/nature04226.
[PMID: 16255080] [PMCID: 1880871] [DOI: 10.1038/nature04226]
[PMID: 16255080] [PMCID: 1880871] [DOI: 10.1038/nature04226]
7)
. Searching for genetic determinants in the new millennium. Nature. 2000 Jun 15;405(6788):847-56. doi: 10.1038/35015718.
[PMID: 10866211] [DOI: 10.1038/35015718]
[PMID: 10866211] [DOI: 10.1038/35015718]
8)
. Balancing life-style and genomics research for disease prevention. Science. 2002 Apr 26;296(5568):695-8. doi: 10.1126/science.1071055.
[PMID: 11976443] [DOI: 10.1126/science.1071055]
[PMID: 11976443] [DOI: 10.1126/science.1071055]
9)
. Dissecting complex disease: the quest for the Philosopher's Stone?. Int J Epidemiol. 2006 Jun;35(3):562-71. doi: 10.1093/ije/dyl001. Epub 2006 Mar 15.
[PMID: 16540539] [DOI: 10.1093/ije/dyl001]
[PMID: 16540539] [DOI: 10.1093/ije/dyl001]
10)
. Genomic priorities and public health. Science. 2003 Oct 24;302(5645):599-601. doi: 10.1126/science.1091468.
[PMID: 14576422] [DOI: 10.1126/science.1091468]
[PMID: 14576422] [DOI: 10.1126/science.1091468]
11)
, 14)
, 20)
. The balance between heritable and environmental aetiology of human disease. Nat Rev Genet. 2006 Dec;7(12):958-65. doi: 10.1038/nrg2009.
[PMID: 17139327] [DOI: 10.1038/nrg2009]
[PMID: 17139327] [DOI: 10.1038/nrg2009]
12)
. Shattuck lecture--medical and societal consequences of the Human Genome Project. N Engl J Med. 1999 Jul 1;341(1):28-37. doi: 10.1056/NEJM199907013410106.
[PMID: 10387940] [DOI: 10.1056/NEJM199907013410106]
[PMID: 10387940] [DOI: 10.1056/NEJM199907013410106]
15)
. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010 May 27;465(7297):473-7. doi: 10.1038/nature09004.
[PMID: 20505728] [DOI: 10.1038/nature09004]
[PMID: 20505728] [DOI: 10.1038/nature09004]
16)
. Cancer genetics. Nature. 2001 May 17;411(6835):336-41. doi: 10.1038/35077207.
[PMID: 11357140] [DOI: 10.1038/35077207]
[PMID: 11357140] [DOI: 10.1038/35077207]
17)
, 23)
. Infectious causation of disease: an evolutionary perspective. Perspect Biol Med. 2000 Spring;43(3):406-48. doi: 10.1353/pbm.2000.0016.
[PMID: 10893730] [DOI: 10.1353/pbm.2000.0016]
[PMID: 10893730] [DOI: 10.1353/pbm.2000.0016]
18)
. The epigenetic basis of twin discordance in age-related diseases. Pediatr Res. 2007 May;61(5 Pt 2):38R-42R. doi: 10.1203/pdr.0b013e31803c7b98.
[PMID: 17413848] [DOI: 10.1203/pdr.0b013e31803c7b98]
[PMID: 17413848] [DOI: 10.1203/pdr.0b013e31803c7b98]
19)
. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000 Jul 13;343(2):78-85. doi: 10.1056/NEJM200007133430201.
[PMID: 10891514] [DOI: 10.1056/NEJM200007133430201]
[PMID: 10891514] [DOI: 10.1056/NEJM200007133430201]
21)
. H. pylori infection and the development of gastric cancer. Keio J Med. 2002 Dec;51 Suppl 2:63-8. doi: 10.2302/kjm.51.supplement2_63.
[PMID: 12528941] [DOI: 10.2302/kjm.51.supplement2_63]
[PMID: 12528941] [DOI: 10.2302/kjm.51.supplement2_63]
22)
. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006 Jun 15;118(12):3030-44. doi: 10.1002/ijc.21731.
[PMID: 16404738] [DOI: 10.1002/ijc.21731]
[PMID: 16404738] [DOI: 10.1002/ijc.21731]
24)
. Common variants conferring risk of schizophrenia. Nature. 2009 Aug 6;460(7256):744-7. doi: 10.1038/nature08186. Epub 2009 Jul 1.
[PMID: 19571808] [PMCID: 3077530] [DOI: 10.1038/nature08186]
[PMID: 19571808] [PMCID: 3077530] [DOI: 10.1038/nature08186]
25)
, 26)
, 27)
. BAK1 gene variation and abdominal aortic aneurysms. Hum Mutat. 2009 Jul;30(7):1043-7. doi: 10.1002/humu.21046.
[PMID: 19514060] [DOI: 10.1002/humu.21046]
[PMID: 19514060] [DOI: 10.1002/humu.21046]
28)
Proal AD et al. Autoimmune disease and the human metagenome. Metagenomics of the human body 231-75(2010).
29)
. Microbial prevalence, diversity and abundance in amniotic fluid during preterm labor: a molecular and culture-based investigation. PLoS One. 2008 Aug 26;3(8):e3056. doi: 10.1371/journal.pone.0003056.
[PMID: 18725970] [PMCID: 2516597] [DOI: 10.1371/journal.pone.0003056]
[PMID: 18725970] [PMCID: 2516597] [DOI: 10.1371/journal.pone.0003056]
30)
, 37)
. Bacterial infection and semen characteristics in infertile men. Arch Androl. 1995 Jul-Aug;35(1):43-7. doi: 10.3109/01485019508987852.
[PMID: 8554430] [DOI: 10.3109/01485019508987852]
[PMID: 8554430] [DOI: 10.3109/01485019508987852]
31)
. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007 Jul 26;357(4):370-9. doi: 10.1056/NEJMsa066082. Epub 2007 Jul 25.
[PMID: 17652652] [DOI: 10.1056/NEJMsa066082]
[PMID: 17652652] [DOI: 10.1056/NEJMsa066082]
32)
. Donor-acquired sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2002 Mar;19(1):18-24.
[PMID: 12002380]
[PMID: 12002380]
33)
. Identification of bacteria on the surface of clinically infected and non-infected prosthetic hip joints removed during revision arthroplasties by 16S rRNA gene sequencing and by microbiological culture. Arthritis Res Ther. 2007;9(3):R46. doi: 10.1186/ar2201.
[PMID: 17501992] [PMCID: 2206354] [DOI: 10.1186/ar2201]
[PMID: 17501992] [PMCID: 2206354] [DOI: 10.1186/ar2201]
34)
. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011 Jun;43(6):585-9. doi: 10.1038/ng.835. Epub 2011 May 15.
[PMID: 21572417] [PMCID: 3115696] [DOI: 10.1038/ng.835]
[PMID: 21572417] [PMCID: 3115696] [DOI: 10.1038/ng.835]
35)
. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012 Aug 23;488(7412):471-5. doi: 10.1038/nature11396.
[PMID: 22914163] [PMCID: 3548427] [DOI: 10.1038/nature11396]
[PMID: 22914163] [PMCID: 3548427] [DOI: 10.1038/nature11396]
36)
. Estimating a woman's risk of having a pregnancy associated with Down's syndrome using her age and serum alpha-fetoprotein level. Br J Obstet Gynaecol. 1987 May;94(5):387-402. doi: 10.1111/j.1471-0528.1987.tb03115.x.
[PMID: 2437951] [DOI: 10.1111/j.1471-0528.1987.tb03115.x]
[PMID: 2437951] [DOI: 10.1111/j.1471-0528.1987.tb03115.x]
38)
. Perinatally acquired neonatal tuberculosis: report of two cases. Pediatr Pathol. 1992 Sep-Oct;12(5):707-16. doi: 10.3109/15513819209024224.
[PMID: 1437883] [DOI: 10.3109/15513819209024224]
[PMID: 1437883] [DOI: 10.3109/15513819209024224]
39)
. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006 Sep;70(3):830-56. doi: 10.1128/MMBR.00016-06.
[PMID: 16959970] [PMCID: 1594586] [DOI: 10.1128/MMBR.00016-06]
[PMID: 16959970] [PMCID: 1594586] [DOI: 10.1128/MMBR.00016-06]
40)
Tost, J. (2008). Epigenetics. Norfolk, UK: Caister Academic Press.
41)
. Hereditary/familial versus sporadic prostate cancer: few indisputable genetic differences and many similar clinicopathological features. Eur Rev Med Pharmacol Sci. 2010 Jan;14(1):31-41.
[PMID: 20184087]
[PMID: 20184087]
home/alternate/genetic_predisposition.txt · Last modified: 09.14.2022 by 127.0.0.1
© 2015, Autoimmunity Research Foundation. All Rights Reserved.